

前編では、マテリアルズインフォマティクス(MI)における計算データセットの役割と、ハイスループット計算の基本的な考え方について解説しました。既存の公開データセットや基盤モデルが充実しつつある現在においても、課題に特化した計算データを自ら生成することは、MIの性能や適用範囲を大きく広げる可能性を秘めています。
後編では、これらの考え方を踏まえたうえで、どのようにしてハイスループット計算を実行してデータ生成を行うか、といった実践的な側面に焦点を当てて解説していきます。ワークフロー管理システムを用いたハイスループット計算の具体的な実装例を通して、計算データ生成を実務に取り入れるためのヒントを示していきます。最後にNECにおける計算データ生成関連の研究をご紹介します。
ハイスループット計算のはじめ方
ここからはハイスループット計算に関するより実践的な話題にフォーカスしていきましょう。まずはハイスループット計算のはじめ方について解説していきます。
ハイスループット計算を始めるためには何を実装すればよいかというと、少なくとも次の3つの処理:入力ファイル作成、計算エンジン(DFT or MDコード)実行、出力ファイル解析を自動で実行できる機能が必要です。加えて、できればデータをデータベースに蓄積する機能と連動しているとよりデータハンドリングが容易になります。
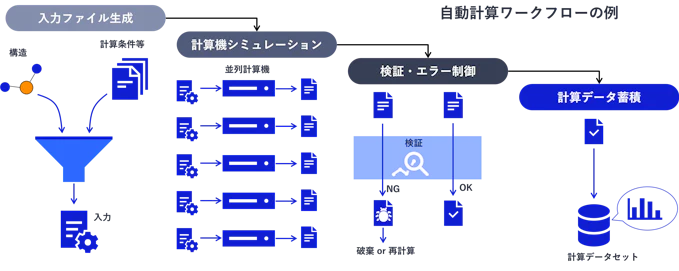
図1. 計算ワークフローの基本構造
ところが、計算化学系の計算エンジンの入出力フォーマットは非常に多様なうえに実行手順が複雑なものも多いため、スクリプト越しで計算を回すことに習熟していないと自動化するのは難しいです。したがって、多くの場合はこれらが既に作りこまれている既存ツール/ソフトウェアの助けを借りるのが現実的です。
ハイスループット計算環境を構築する場合、大きく分けて2通りの選択肢があります。ひとつは、既存の計算自動化ツールを使うことです。近年は様々な専用ツールが開発されており、無償で公開されているものも多いです。それらを利用することで、手軽にハイスループット計算を実践することができます。できる計算の種類や使用法は完全にツールに依存してしまいますが、目的と合致するツールが見つかれば簡単にハイスループット計算をはじめることができるでしょう。以下に、オープンソースソフトウェアとして公開されているハイスループット計算ツールのうちメジャーなものをいくつか挙げてみました。
ツール名 | 計算手法* | 対象系 | 扱える物性の例 |
DFT | 固体、分子 | バンド構造、フォノン | |
DFT | 固体 | 構造安定性、Bader電荷 | |
DFT, HF | 分子 | エネルギー、分極 | |
MD | 高分子 | ガラス転移温度、熱伝導度 | |
MD | 生体分子 | 自由エネルギー、結合親和性 | |
DFT, TB | 金属錯体 | スピン分裂エネルギー | |
DFT | 合金 | 熱力学状態図 |
(* DFT=密度汎関数法, MD=分子動力学, HF=Hartree-Fock法, TB=強結合法)
他にも様々なソフトウェアがあるので、興味のある計算手法について調べてみてください。いくつか見比べてみて使いやすそうなものから試してみるのが良いと思います。また、シミュレーションそのものの経験が無ければ、技術的なサポートを受けられる有償ソフトウェアまたはサービスを利用するという手もあります。
もうひとつはワークフロー管理システム (WMS: Workflow Management System) を導入して自分で設計する方法です。WMSを利用したハイスループット計算の特徴はワークフロー設計の自由度が高いことで、大抵のソフトウェアは任意の計算ワークフローに対応できるように作られています。そのため既存の自動化ツールに実装されていないような計算タスクを実行できたり、自作コードを組み込めたり柔軟な対応が可能です。また、自由度の高さを活かすことで、データ生成効率を向上させるための工夫(エラー制御、リソース割当て最適化、等)を盛り込みやすい、というのも特筆すべき利点です。その一方で、多少のワークフロー設計は必要なのでハイスループット計算を開始するまでに若干準備を要するというのが欠点です。また、システム設計に関する専門性は不要ですが、システムを円滑に運用するためには多少の習熟がいるかもしれません。自由度の高さと習得コストはある程度トレードオフになるため、既存の自動化ツールをそのまま使えるのかMWSでの設計が必要か、状況に応じた使い分けが大事です。
データ生成効率を追求するためには計算の自動化だけではなく、エラー制御やリモート計算資源との通信、パフォーマンス情報等のメタデータの取得といった様々な機能が要求されます。加えて、生成される大量のデータ/メタデータを適切に処理・蓄積するためのデータ管理機能も重要になってきます。WMSはこういった多彩な機能を担う複合的なシステムです。汎用のものを含めると様々なWMSが開発されていますが、ハイスループット計算を指向して設計されているものとしては”AiiDA”や”FireWorks”があります。これらのWMSを上手く活用することで、情報システムの専門家でなくても高度な計算管理機構をハイスループット計算に組み込むことができます。
ワークフロー管理システム AiiDAで実践するハイスループット計算
百聞は一見にしかず、ということでハイスループット計算の実行例をいくつか見てみましょう。
ここでは先に挙げたWMSのひとつ “AiiDA” を使って計算ワークフローを設計します。AiiDAは欧州のグループを中心に開発されているPythonベースのWMSで、オープンライセンス(MITライセンス)で利用可能です。選定の理由は、次にまとめるようにハイスループット計算に望ましい機能を一通り備えており、かつ開発者コミュニティの規模が大きく(本稿執筆時点で106名)開発が活発なためです。
AiiDAの主要な機能は以下のようにまとめられます。
- データ来歴の補足:ワークフロー実行中のデータ来歴(データの出自および変換の記録)を逐一メタデータとして記録する機構を備えており、結果に至るまでの入力・処理・条件を一貫して記録することで、再現性・信頼性・追跡可能性を担保します。オープンデータの公開方法に関する原則(FAIR原則※)に基づいて設計された機能であり、堅牢かつ安全なデータ管理を行えます。
- 動的なワークフロー実行管理:ワークフローの実行時制御に対応しており、高度な条件分岐処理やエラー制御が可能です。また、適応的なスケジューリングやリソース割り当ても可能です。
- 階層的なワークフロー構成:ワークフローから別のワークフローを呼び出す、といった階層的なワークフロー構成に対応しています。これにより、大規模なアプリケーションの開発が容易になり、高いモジュール性と再利用性を実現できます。
- リモート計算資源との通信:ローカル環境だけではなく、リモート計算資源上での計算ジョブの実行および監視にも対応しています。これにより計算資源を必要に応じて容易に増強でき、特に大型計算機やクラウド計算資源を使用する際に便利です。
- Pythonエコシステム:Python APIを完備しており、Pythonが使えれば全ての機能にアクセスできます。そのためスクリプト実行だけでなく対話シェルやノートブックなどPythonに対応するインタフェースを一通り利用できます。ワークフローはモジュール化してパッケージとして管理できるため、ワークフローのインポート・エクスポートが容易にできます。
このようにWMSとして有用な機能を備えています。中でも①のデータ来歴補足機能を備えている点が特徴的で、近年は欧州を中心に材料データにおいてもFAIR原則の普及が進んでおり、組織間データ連携やオープンサイエンスの文脈で注目を集めています。
※FAIR原則:研究データを「見つけやすく(Findable)」「アクセス可能(Accessible)」「相互運用可能(Interoperable)」「再利用可能(Reusable)」に整備し、研究者間の効率的な共有と再利用を促進するための指針です。
前置きが長くなりましたが、ここからいくつか実行例をご紹介していきます:
シングルショット計算―バンドギャップ
最も単純なハイスループット計算ワークフローとして、単発の電子状態計算から得られる量、例えばバンドギャップ (Eg) を計算するとします。結晶構造データベースから適当にサンプリングした構造について、DFTによる電子状態計算を実行して出力結果を取得する、すなわち下図のような計算ワークフローを構造の数だけ実行します。最終的に計算結果からバンドギャップの値を読み出すことで、入力構造とバンドギャップのペアからなるデータセットが形成されます(図では値を集約して金属、半導体、絶縁体に分類した結果を示しています)。これがハイスループット計算による計算データセット生成の最小構成で、最も基本となる形式です。バンドギャップ以外にも、全エネルギーや磁気モーメントなどのデータ生成を行う場合も同様です。
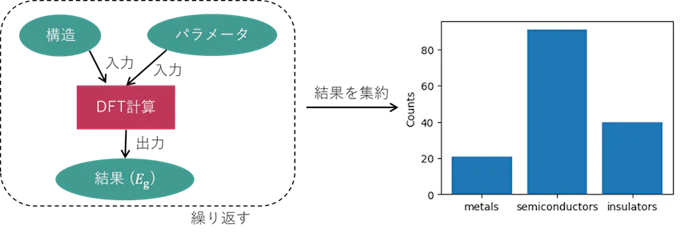
図2. シングルショットDFT計算と結果分布
マルチショット計算―関数フィッティング
シングルショット計算の例はワークフローというには単純すぎるので、次はよりワークフローらしい例として多段の計算プロセスがからむ課題を見てみましょう。ここではDFT計算の結果を用いて、等方性固体の状態方程式への関数フィッティングを行います。
状態方程式は全エネルギーを体積の関数として記述する式、つまり Etot(V) なので、体積を変化させながら全エネルギーをDFTによって計算するという手順を複数回行う必要があります。このような処理を行うワークフローを下図の左側に示しました。まず入力となる母構造を膨張/圧縮して体積変化した構造を複数生成し(2段目四角の処理)、各構造に対してDFT計算を実行(4段目四角の処理)、各体積における全エネルギーの値を用いてフィッティングを行い(6段目四角の処理)最終的にフィッティングパラメータを得ることができます。
このような処理を様々な物質に対して行った結果が右の図で、それぞれの曲線が異なる物質に対応しています。つまり一本の曲線を得るのに左図のような処理を行う必要があるため、(できなくはないかもしれませんが)手動で行うのは少々面倒です。このように複数のプロセスからなるハイスループット計算においては、ワークフローを組んだ上で自動実行するのが効率的です。ちなみに、状態方程式フィッティングから得られる情報として、右図の曲線の極小値が平衡体積、曲率がバルク弾性率に対応します。
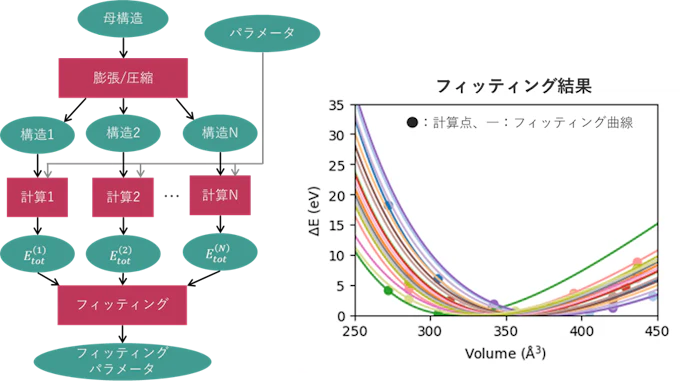
図3. マルチショット計算と状態方程式フィッティング
制御構造を有するワークフロー―立体配座探索
これまでの例では入力構造はあらかじめ用意されたものを読み込んでいましたが、最後にWMSのありがたみが分かる例として、制御構造を有する複雑なワークフローを実行してみましょう。固体の例が続いたので、趣を変えて分子の立体配座探索を実行してみます。
回転可能な結合を持つ分子は様々なパターンの立体配座を取り得るため、その中から安定構造を探索する立体配座探索が重要になります。しかしながら、現在広く使われている構造最適化手法は初期配置に依存するため、複数の初期構造を用意する必要があります。流れとしては下図左のワークフローのようになりますが、少々煩雑なので主に図右のフローチャートを使って説明します。入力としては構造式をSMILES文字列などの形式で与えます。与えられた構造式に対して、構造サンプリングアルゴリズムまたは生成モデルなどで立体配座の候補を複数生成し、各候補に対して第一原理計算による構造最適化を行います。その後、各候補からエネルギーが最も低い構造を最安定構造とし、必要であればポストプロセス計算による物性値の評価などを行います。
このワークフローにおいてハイスループット計算の観点から重要なのは、候補配座に関する反復や最安定構造の取捨選択といった制御が必要ということです。上でお見せしたマルチショット計算の例ではワークフローの構造は固定でしたが、こちらは入力や中間出力によって実行するワークフローの構造が適応的に変化します。このような複雑な処理でも、AiiDAのようなWMSを使用すれば難なく自動化することができます。
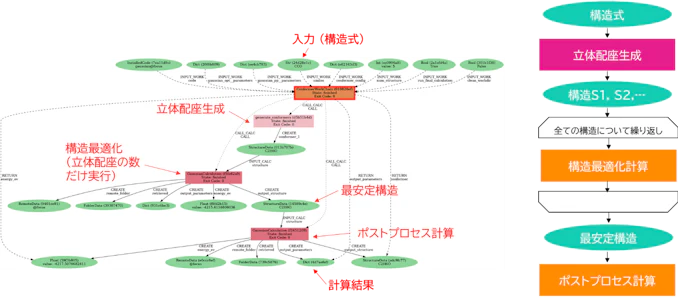
図4. 制御構造を持つ配座探索ワークフロー
今回は実行例をお見せするだけでしたが、実際にAiiDAを使ってみたい場合はチュートリアル記事をQiitaにて連載しておりますので、興味がありましたらそちらもご参照ください。
なお、AiiDA以外にも科学向けのWMSは数多く存在しています。主要なWMSとその機能などを体系的にまとめたレビューがあるので、もしWMSの選定から行いたい場合はこちらの文献が参考になるかと思います。
計算×MIに関するNECの取り組みのご紹介
最後に、わたしたちNECのMIに関する取り組みのうちハイスループット計算に関連するものをいくつか簡単にご紹介いたします:
ハイスループット物性計算
第一原理計算と物性理論を組み合わせたハイスループット計算を行うことで、単発の計算では評価できない様々な物性量についてデータセット生成を行っています。図の例では近年スピントロニクス分野において盛んに研究がなされている現象である異常ホール効果、スピンホール伝導度 (SHC) ・軌道ホール伝導度 (OHC) について、DFTと線形応答理論(久保公式)を組み合わせたハイスループット計算による網羅的評価を行った結果です(参考研究)。
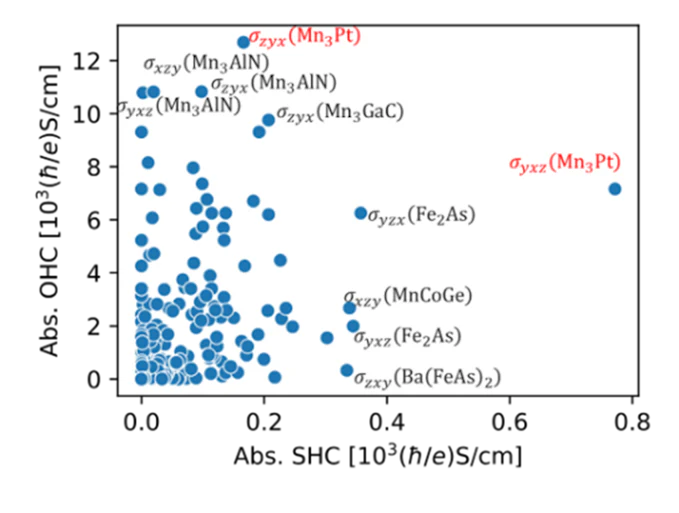
図5. スピントロニクス分野でのハイスループット計算結果
物性計算のベクトル計算機向け最適化
久保公式は計算コストが高く、物性計算ワークフロー全体のボトルネックになっていました。そこでベクトル計算機を活用したHPC技術によって久保公式を高速化し、全体のデータ生成スループットを改善する手法を開発しました。数値積分ループ内の行列演算をバッチ処理することでベクトル演算器の使用率を高め、従来の実装(CPU+MPI並列)よりも高い計算効率を実現できました。

図6. 久保公式計算ワークフローと所要計算時間
自律材料探索
第一原理計算と機械学習を組み合わせることで、材料開発を完全に自動で行える自律探索システムを実現しました。材料開発は合成、評価、実験計画のステップを繰り返すことで行われますが、合成と評価を第一原理計算、実験計画を機械学習によって代替することで仮想空間(コンピュータ)内に閉じたループを構成することができ、人による入力を介さずに自律的に材料探索を行うことができます。実際にこの手法を磁性材料探索に活用することで、最終的に高い性能を持つ磁性合金の発見に成功しました。
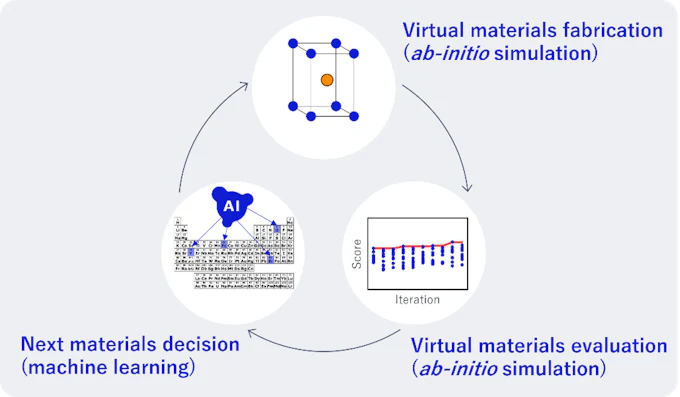
図7. 第一原理計算と機械学習を統合した閉ループ型の自律材料探索プロセス
能動学習による機械学習原子間ポテンシャル学習
機械学習原子間ポテンシャル (MLIP) を能動学習によって効率よく作成する手法・コードを開発しています。不確実性の大きな(=学習の足りていない)配位を効率的にサンプリングできるよう工夫されており、特異的な原子配置に対しても頑健なMLIPを構築できます。本手法を自由エネルギー計算に応用し、溶質―溶媒系の過剰化学ポテンシャルを従来手法よりも高精度に算出できることを実証しました。
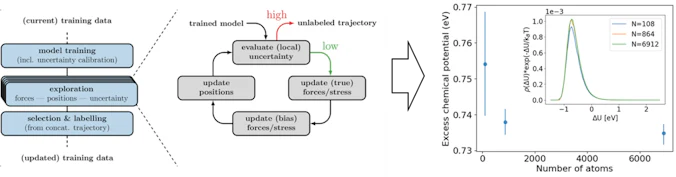
図8. 能動学習による機械学習原子間ポテンシャルの高効率構築
計算から実験への転移学習 (Sim2Real転移学習)
計算データの最大の弱点は、近似や単純化に由来する現実との系統的なズレです。そこで計算から実験への転移学習を行うことで、計算データの数の多さを活かして安定した学習を行いつつ、実験データによって現実とのズレを補正する、というデータ生成コストと精度を両立した学習法を開発しています。本手法を電極触媒の活性予測に適用し、転移学習によって実験データが少量でも高性能なモデルが学習できることを実証しました。
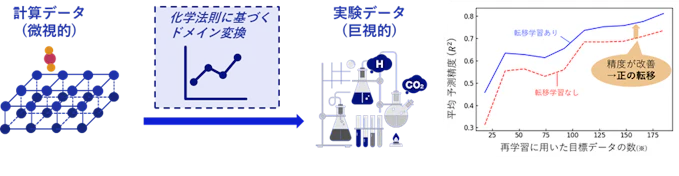
図9. Sim2Real転移学習の概念図
おわりに
今回の記事では、マテリアルズインフォマティクス(MI)における計算データセットの役割について議論し、ハイスループット計算の基礎から具体的な実践例まで解説いたしました。長くなってしまいましたが、少しでもご参考になりましたら、そしてハイスループット計算をやってみたい気持ちになって頂けましたら幸いです。ここまでお読みいただきありがとうございました。
参考文献
- atomate2 (https://materialsproject.github.io/atomate2/) 論文: A. M. Ganose et al., "Atomate2: modular workflows for materials science", Digital Discovery 4, 1944–1973 (2025).
- AFLOW (https://aflow.org/) 論文: S. Curtarolo et al., "AFLOW: An automatic framework for high-throughput materials discovery", Comput. Mater. Sci. 58, 218–226 (2012).
- MolSSI QCArchive (https://docs.qcarchive.molssi.org/) 論文: D. G. A. Smith et al., "The MolSSI QCArchive project: An open-source platform to compute, organize, and share quantum chemistry data", WIREs Comput. Mol. Sci. 11, e1491 (2021).
- RadonPy (https://github.com/RadonPy/RadonPy) 論文: Y. Hayashi et al., "RadonPy: automated physical property calculation using all-atom classical molecular dynamics simulations for polymer informatics", npj Comput. Mater. 8, 222 (2022).
- BioSimSpace (https://biosimspace.openbiosim.org/) 論文: L. O. Hedges et al., "BioSimSpace: An interoperable Python framework for biomolecular simulation", J. Open Source Softw. 4(43), 1831 (2019).
- molSimplify (https://github.com/hjkgrp/molSimplify) 論文: E. I. Ioannidis, T. Z. H. Gani, H. J. Kulik, "molSimplify: A Toolkit for Automating Discovery in Inorganic Chemistry", J. Comput. Chem. 37, 2106–2117 (2016).
- Alloy Theoretic Automated Toolkit (ATAT) (https://axelvandewalle.github.io/www-avdw/atat/) 論文: A. van de Walle, M. Asta, G. Ceder, "The Alloy Theoretic Automated Toolkit: A user guide", Calphad 26, 539–553 (2002).
- AiiDA (https://www.aiida.net/) 論文: S. P. Huber et al., "AiiDA 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance", Sci. Data 7, 300 (2020); M. Uhrin, et al., “Workflows in AiiDA: Engineering a high-throughput, event-based engine for robust and modular computational workflows”, Comput. Mater. Sci. 187, 110086 (2021).
- FireWorks (https://materialsproject.github.io/fireworks/), 論文: S. P. Ong et al., "FireWorks: a dynamic workflow system designed for high-throughput applications", Comput. Mater. Sci. 93, 209–215 (2014).
- G. Pizzi, et al., “AiiDA: automated interactive infrastructure and database for computational science”, Comput. Mater. Sci. 111, 218 (2016).
- 1FORCE11, “THE FAIR DATA PRINCIPLES” (2016). https://www.force11.org/group/fairgroup/fairprinciples, NBDC研究チーム(訳), "FAIR原則(「THE FAIR DATA PRINCIPLES」和訳)" (2019)
- AiiDAではじめる計算自動化- Qiita (https://qiita.com/yuta-yahagi/items/92d907b46fcc10eab9e6).
- F. Suter et al., “A terminology for scientific workflow systems”, Future Gener. Comput. Syst. 174, 107974 (2026).
- J. Železný, et al., High-throughput study of the anomalous Hall effect, Npj Comput. Mater. 9, 1 (2023); Y. Yahagi and J. Železný, "High-throughput study of spin and orbital transport in magnetic materials", MMM2022, Minneapolis (2022)
- 矢作裕太、加藤季広, 『量子輸送特性計算法のベクトル計算機向け最適化』, 研究報告 ハイパフォーマンスコンピューティング(HPC) 2024-HPC-194, 1 (2024).
- Y. Iwasaki, R. Sawada, E. Saitoh, and M. Ishida, “Machine learning autonomous identification of magnetic alloys beyond the Slater-Pauling limit”, Commun. Mater. 2, 1 (2021).
- ALBREW (https://github.com/nec-research/alebrew) 論文:V. Zaverkin, et al., “Uncertainty-biased molecular dynamics for learning uniformly accurate interatomic potentials”, Npj Comput. Mater. 10, 83 (2024).
- 澁谷泰蔵、Zaverkin Viktor、矢作裕太、Alesiani Francesco、『機械学習ポテンシャルを用いた粒子挿入法による液体金属の自由エネルギー計算』第39回 分子シミュレーション討論会、松江 (2025)
- Y. Yahagi, K. Obuchi, F. Kosaka, and K. Matsui, “Transfer learning from first-principles calculations to experiments with chemistry-informed domain transformation”, Mach. Learn. Sci. Technol. 6, 025026 (2025).















